
Как разрабатывают лекарства
Примерно до середины XX века лекарства чаще всего обнаруживали случайно: достаточно вспомнить счастливую историю открытия Яном Флемингом пенициллина. Раньше у учёных не хватало знаний о молекулярных механизмах заболеваний, поэтому невозможно было создавать молекулы, прицельно влияющие на них.
В XX веке наука совершила огромный рывок в области химии и физики, который привёл и к трансформации биологии и медицины. Была создана молекулярная биология, разработано множество методов анализа и синтеза сложных молекул. К 1990-м годам это привело к новым подходам, когда поиск лекарств с самого начала сфокусирован на молекулярных механизмах развития заболеваний и направлен на ключевые молекулы, отвечающие за их развитие – молекулярные мишени. Такой подход называется рациональной разработкой, а лекарства – таргетными (от англ. target – цель, мишень). Как правило, мишень в организме - это белок, играющий важную роль в патогенезе заболевания. Ещё до начала разработки нового лекарства проводится большая работа по выяснению того, на какие белки нужно повлиять (заблокировать, усилить или ослабить функцию), чтобы изменить ход заболевания. Эта деятельность ведется в академических учреждениях: университетах, научно-исследовательских институтах, медицинских центрах, но такими исследованиями занимаются и крупные фармкомпании. Когда мишень выбрана, начинается поиск лекарств, воздействующих на нее.
Как можно классифицировать лекарства?
До 1990-х годов основным типом лекарственных молекул были низкомолекулярные вещества – их обычно легче синтезировать, анализировать, с ними проще работать. Они и сейчас остаются самыми распространенными – достаточно зайти в любую аптеку.
Начиная с 2000-х годов на арену выходят биопрепараты, в первую очередь моноклональные антитела. Их применяют преимущественно для лечения тяжелых заболеваний, например, онкологических и аутоиммунных. Биопрепараты более дорогие и сложные в производстве, но они позволяют добиться такой высокой избирательности (а значит безопасности) и эффективности, что прочно заняли лидирующее место и в арсенале медицины, и в рейтингах продаж.
Наконец, третье поколение лекарств только начинает свой путь. Это самые передовые препараты – генная и клеточная терапия. Они способны не только уменьшить симптомы или замедлить развитие заболеваний, но и в ряде случаев привести к полному излечению. Их развитие стало возможным благодаря более глубокому понимаю взаимодействий между клетками, прохождения сигналов внутри клеток, совершенствованию методов генной инженерии и не в последнюю очередь – скачку в компьютерных технологиях. Пока препаратов передовой терапии очень немного, и они страшно дорогие (стоимость некоторых за курс составляет около 1 млн. долларов США), но есть надежда, что ситуация изменится с развитием автоматизации, появлением новых методов молекулярной и клеточной биологии.
Стадии разработки лекарств
![Источник информации: DiMasi et al. [2016], Wong et al. [2018]](https://opharme.ru/storage/uploads/publication/94/1640871531_InfoGraphs_14 1.jpg)
Этап I. Открытие лекарств
Процесс открытия лекарства напоминает очень широкую воронку, в которую попадают сотни тысяч структур, смоделированных компьютером, а на выходе остается одна лучшая молекула, экспериментально проверенная в лаборатории. Этап открытия проходит чаще всего внутри фармкомпаний: в R&D отделах бигфармы или в маленьких стартапах, специально созданных для воплощения новой терапевтической идеи. По времени процесс может растянуться на годы, но иногда компаниям удается пройти его и за несколько месяцев. Стоит такая разработка обычно не очень дорого: стартапу может хватить 1-5 млн долларов.
Поиск начинается с компьютерных расчётов (эта стадия называется in silico – от лат. «в кремнии», то есть в «мозге» компьютера). Методы молекулярного моделирования, использование информации о строении предыдущих лекарств в сочетании с алгоритмами машинного обучения позволяют предсказать структуры веществ, которые хорошо свяжутся с мишенью. Таких структур может получиться очень много, сотни тысяч. Но на практике не все компьютерные предсказания сбудутся: следом наступает этап экспериментальной проверки – in vitro (от лат. «в стекле»). Сначала в ходе скрининга отбрасываются заведомо «плохие» кандидаты: те, которые не растворяются, не связываются с мишенью или недостаточно стабильны. Остаются десятки кандидатов (их называют «хиты»), на основе которых получают новые версии молекул – «лиды». Лиды также проходят тестирование in vitro на связывание с мишенью и на различные фармакологические свойства. Например, уже на этапе тестирования можно определить, не будет ли вещество слишком быстро выводиться из организма или отравлять его. Наконец, из нескольких лидов выбирается самый лучший: он становится лекарственным кандидатом. На этом заканчивается этап drug discovery («открытие лекарства») и начинается drug development («разработка лекарства»).
Этап II. Доклинические исследования
Первая стадия этапа drug development называется ДОклиническая, потому что она проводится до клинических экспериментов, то есть до экспериментов на людях. Она включает в себя тестирование на клетках (которое тоже называется in vitro) и на животных (in vivo, от лат. «в живом»). На этом этапе ученые пытаются получить подробную информацию о безопасности и эффективности лекарственного кандидата. Для этого используют модели заболеваний, то есть пытаются в условиях in vitro или in vivo имитировать какие-то аспекты болезней человека. Не всегда удается это сделать с достаточной надёжностью, но совсем неудачных кандидатов отсеять возможно. Не факт, что лекарство, которое помогло мыши, поможет и человеку, но если уж даже мышь оно вылечить не в состоянии, то и человека вряд ли вылечит. В качестве модельных используют животных, у которых механизм развития фокусного заболевания наиболее похож на аналогичный у человека. Это могут быть мыши, крысы, собаки, свиньи, кролики, обезьяны. Для каждой болезни выработаны свои подходы.
После того, как лекарственный кандидат показал приемлемую эффективность и безопасность в моделях, наступает последний этап доклинического тестирования на токсичность. Начиная с этой стадии, к качеству проведения экспериментов предъявляются очень строгие требования, потому что от него зависит безопасность людей, которые будут получать лекарство на следующем клиническом этапе. За выполнением требований следят государственные органы (например, Минздрав в России, FDA в США, ЕМА в ЕС), которые тщательно изучают досье при подаче на следующий этап – клинические исследования.
Этап III. Клинические исследования
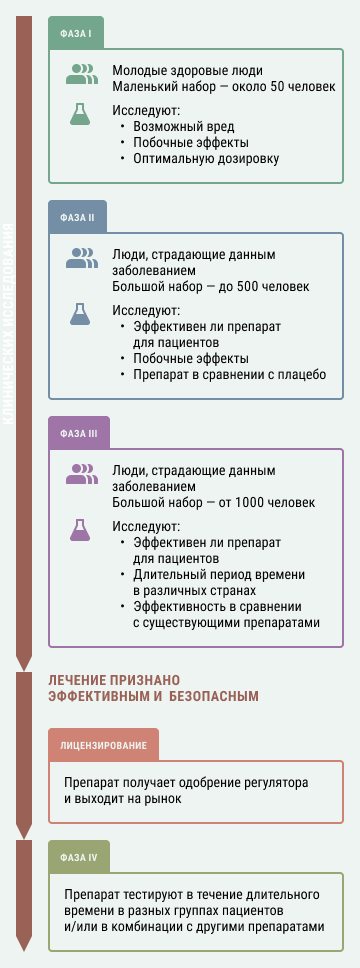
Цель клинических исследований – не «доказать эффективность и безопасность» лекарства, как это часто представляют. Безопасных лекарств не бывает: если вещество воздействует на организм, то риск нежелательных явлений есть всегда (это в не меньшей степени относится и к так называемым натуральным препаратам на основе лекарственных растений). Их цель - сделать вывод о соотношении пользы и рисков при лечении конкретной болезни. Для разных болезней – разные требования. Например, к спреям от насморка или любым вакцинам применяются высочайшие требования по безопасности – они вводятся миллионам практически здоровых людей, поэтому недопустим даже минимальный уровень тяжелых нежелательных явлений. А вот к лекарствам для лечения онкологических больных в поздней стадии требования гораздо мягче. За полное выздоровление (а сейчас такое бывает все чаще) вполне можно заплатить даже временной госпитализацией из-за нежелательных явлений лекарства. Хотя, конечно, ученые пытаются делать препараты всё безопаснее и эффективнее, а индустрия охотно финансирует такие разработки, потому что они точно будут пользоваться спросом.
Чаще всего клинические исследования нового лекарства делят на три фазы. На первой фазе лекарство получает небольшое количество добровольцев (обычно 10-50 человек), зачастую это здоровые люди, если риск токсичности не велик. Цель первой фазы – изучение безопасности, фармакокинетики (изучают превращения лекарства в организме) и фармакодинамики (изучают действие лекарства на организм) вещества. Во второй фазе участвуют уже обязательно больные данным заболеванием - пациенты (обычно 50-200 человек). Исследователи продолжают изучать безопасность, получают первые данные об эффективности в различных дозировках и режимах введения. Во второй фазе может применяться дизайн с параллельными группами, которые получают плацебо. Вторая фаза или фазы (их может быть несколько) служат в том числе для того, чтобы правильно запланировать третью – ключевую.
Классические исследования третьей фазы – двойные слепые (рандомизированные,когда пациент не знает, что он получает) контролируемые – стандарт тестирования лекарств в области доказательной медицины. В качестве группы контроля возможно использование плацебо, однако если на рынке уже есть стандарт терапии - эффективный препарат для лечения данного заболевания, то сравнивать новое лекарство необходимо с ним. В исследовании III фазы проверяют заранее сформулированную гипотезу об эффективности лекарства (например, «лекарство у данных пациентов по такому-то показателю на 20% эффективнее стандартной терапии») и получают расширенные данные о безопасности. Число пациентов - участников III фазы может быть от нескольких сотен до многих тысяч.
Необходимо понимать, что клинические исследования – это научный эксперимент с участием людей. Поэтому по отношению к их участникам применяются принципы Хельсинской декларации, главные из которых – добровольность и информированность.
Когда лекарство начинают тестировать на людях, как и в любом научном эксперименте, результат заранее не известен. По статистике, успешно проходят все три фазы исследований лишь 10-15% препаратов. Сейчас новые технологии машинного обучения, огромные массивы электронных данных, новые методы анализа и визуализации биологических процессов дают надежду на то, что этот процент удастся повысить. Было бы здорово научиться на ранних этапах отсекать бесперспективные разработки и пускать в исследования на людях только те, что будут действительно эффективными, а то и вообще заменить часть клинических исследований моделированием – это позволило бы не подвергать риску участников исследований, снизить сроки и затраты на разработку новых лекарств. Однако пока биология - слишком крепкий орешек, клинические исследования ничем нельзя заменить, и неприятные сюрпризы в виде провалов случаются регулярно.
В среднем процесс разработки от получения лекарственного кандидата до выхода на рынок занимает около 10 лет и обходится в сотни миллионов долларов. Однако сейчас всё больше маленьких стартапов выводят на рынок свои разработки, особенно в области орфанных заболеваний, в отношении которых регуляторные требования менее строгие.
Если лекарство оказалось достаточно эффективным и безопасным, компания подает в регуляторный орган досье на одобрение и препарат выходит на рынок. Часто после этого проводят так называемые пострегистрационные исследования четвертой фазы, наблюдая за большими группами людей, получающих лекарство в реальных условиях. Всё-таки тысячи пациентов в контролируемом эксперименте – это не то же самое, что миллионы людей разной расы, возраста, пола, с сопутствующими заболеваниями, параллельно принимающие другие лекарства. Благодаря таким исследованиям можно понять, как действует препарат у самых разнообразных подгрупп пациентов в течение длительного времени, почему одним он помогает лучше, а другим - хуже. Это может дать информацию для изменения клинических рекомендаций по применению препарата или для разработки новых, более эффективных и безопасных.

